فیلوژنی چیست؟
-
مقدمهای بر فیلوژنی – شامل تعریف، تاریخچه و اهمیت آن
-
مفاهیم پایه – شامل اصطلاحات درخت تبارزایی، ریشهگذاری، و گرهها
-
انواع شواهد فیلوژنتیکی – شامل شواهد مورفولوژیکی (ریختشناسی)، مولکولی و ژنومی
-
روشهای ساخت درخت تبارزایی
-
روشهای بر پایه فاصله (مانند Neighbor-Joining)
-
روشهای بر پایه ویژگی (مانند Maximum Parsimony و Maximum Likelihood)
-
استنتاج بیزی (Bayesian Inference)
-
-
ساعتهای مولکولی و تعیین زمان واگرایی گونهها
-
فیلوژنومیک و دادههای پرحجم (High-Throughput)
-
مفاهیم گونه و ارتباط آن با فیلوژنی – شامل گونههای زیستی، فیلوژنتیکی و غیره
-
کاربردها – در طبقهبندی زیستی، حفاظت از گونهها، پزشکی، اپیدمیولوژی
-
نرمافزارها و ابزارهای محاسباتی – مانند MEGA، RAxML، BEAST و دیگر ابزارهای تحلیلی
-
محدودیتها، خطاهای رایج و روشهای بهینه
-
مطالعات موردی – درختهای معروف مربوط به مهرهداران، ویروسها و گیاهان
-
مسیرهای آینده – شامل روشهای نوین، دادههای بزرگ (Big Data) و ادغام دیرینهشناسی با تبارزایی
فیلوژنی چیست؟
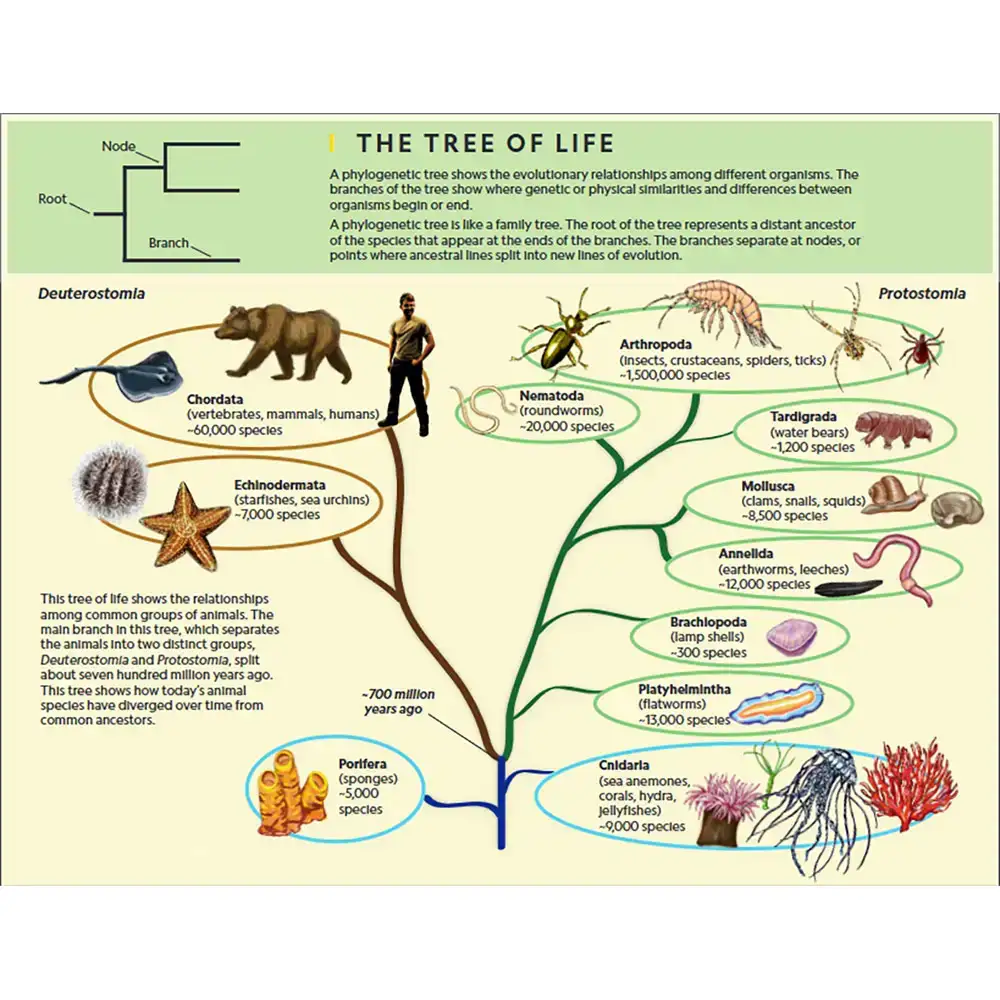
فیلوژنی (Phylogeny) به مطالعه تاریخچه تکاملی و روابط بین موجودات زنده یا گروههایی از موجودات اشاره دارد. این روابط معمولاً به شکل یک نمودار شاخهای به نام درخت فیلوژنتیک (Phylogenetic Tree) نمایش داده میشوند؛ نموداری که نشان میدهد گونهها یا گروهها چگونه در طول زمان از نیاکان مشترک خود جدا شده و تکامل یافتهاند.
فیلوژنی به یکی از عمیقترین پرسشهای زیستشناسی پاسخ میدهد:
«همه موجودات زنده چگونه به هم مرتبط هستند؟»
این علم، الگوهای نیاکان مشترک و واگرایی را آشکار میکند و نشان میدهد که موجودات چگونه از ریشههای مشترک، منشأ گرفته و گوناگون شدهاند.
فیلوژنتیک (Phylogenetics)
فیلوژنتیک یعنی مطالعهی فیلوژنی یا بررسی روابط تکاملی میان گونهها و گروهها از طریق بررسی توالی ژنتیکی آنها.
فیلوژنتیک، بازوی علمی سامانهبندی زیستی (سیستماتیک) و ردهبندی (تاکسونومی) است. امروزه، روابط فیلوژنتیکی بیشتر از طریق توالییابی DNA تعیین میشود، که پلی میان زیستشناسی مولکولی و ردهبندی سنتی ایجاد کرده است.
این مسئله باعث شده که دانشمندان میدانی و محققان آزمایشگاهی مجبور شوند با هم ارتباط برقرار کنند، که پدیدهای مثبت و نوین محسوب میشود.
همچنین، فیلوژنی این امکان را میدهد که نیاکان مشترک فرضی بین موجودات شناسایی شوند. اگر درخت فیلوژنتیک دارای چنین نیاکانی باشد، به آن "درخت ریشهدار" (Rooted Tree) میگویند.
مثالی از کاربرد فیلوژنتیک: کشف اینکه بیشتر یوکاریوتهای فتوسنتزکننده (گیاهان و جلبکها) به هم مرتبط هستند و احتمالاً نیاکان مشترک داشتهاند.
اما درختهای فیلوژنتیک میتوانند توسط انتقال افقی ژن (HGT) به شدت دچار انحراف شوند. HGT یعنی انتقال ژن بین گونههایی که مستقیماً با هم خویشاوند نیستند.
این پدیده میتواند تصویری نادرست از روابط فیلوژنتیک بین گونهها ارائه دهد.
فیلوژنومیکس (Phylogenomics)
اصطلاح "فیلوژنومیکس" توسط جاناتان آیزن (Jonathan Eisen) در سال ۱۹۹۸ ابداع شد تا به محدودیتهای مطالعههای تکاملی مبتنی بر توالی ژنها پاسخ دهد.
او معتقد بود که شباهت توالی ژنی لزوماً به معنی شباهت عملکردی آنها نیست.
وی گفت:
«پیشبینی عملکرد ژنها زمانی بسیار دقیقتر میشود که بهجای تمرکز صرف بر شباهت توالی، روی نحوهی تکامل آن ژنها تمرکز کنیم.»
فیلوژنومیکس، مطالعهی فیلوژنتیک را که معمولاً با توالی یک ژن خاص کار میکند، گسترش داده و به بررسی کل ژنوم میپردازد.
این واژه در معانی مختلفی استفاده میشود، اما همهی کاربردهای آن به نوعی شامل ترکیب ژنومیک با تکامل است.
مثالی از کاربرد فیلوژنومیکس در میکروبیولوژی: بررسی جنس دریایی Salinospora.
در مجموع، فیلوژنومیکس بر مقایسهی کل ژنومها تکیه دارد، در حالی که فیلوژنتیک معمولاً یک یا چند ژن خاص را مقایسه میکند تا به پیشبینی عملکرد ژن برسد.
به همین دلیل، فیلوژنومیکس میتواند:
-
رویدادهای تکاملی درون ژنها مثل تکرارها (دوبلیکیشن) یا حذفها (دلیشن) را شناسایی کند.
-
ژنهایی را که از طریق انتقال افقی وارد یک ژنوم شدهاند، تشخیص دهد.
ریشه واژه و تعریف
واژه phylogeny از دو واژه یونانی گرفته شده است:
-
"phylon" به معنای “قبیله” یا “نژاد”
-
"genesis" به معنای “پیدایش” یا “آفرینش”
بنابراین، فیلوژنی بهمعنای واقعی کلمه یعنی:
«پیدایش و توسعه یک قبیله یا گونه»
تعریف امروزی زیستشناسی از فیلوژنی چنین است:
"توسعه تکاملی و تنوعیابی یک گونه یا گروهی از موجودات زنده، یا تاریخچه تکاملی یک گروه ردهبندیشده زیستی (taxonomic group)."
تفاوت فیلوژنی، تاکسونومی و سیستماتیک
|
واژه |
تعریف |
|
Phylogeny |
تاریخچه تکاملی و روابط بین موجودات زنده |
|
Taxonomy |
علم نامگذاری، توصیف و ردهبندی موجودات زنده |
|
Systematics |
علمی گستردهتر که تاکسونومی و فیلوژنی را برای درک تنوع زیستی و روابط موجودات ترکیب میکند |
فیلوژنی ستون فقرات سیستماتیک محسوب میشود؛ یعنی ساختار شاخهای که ردهبندی علمی موجودات بر اساس آن شکل میگیرد.
تاریخچه توسعه فیلوژنی
۱. دیدگاههای پیش از داروین
پیش از نظریه داروین، طبقهبندیها بر اساس ویژگیهای ظاهری (مورفولوژیک) انجام میشدند و هدفشان بیشتر توصیف شباهتها بود، نه روابط نیاکانی.
-
ارسطو: نخستین نظامهای ردهبندی زیستی را بر پایه ویژگیهای ظاهری طراحی کرد.
-
لینه (قرن ۱۸): نظام طبقهبندی دودویی (دو اسمی) و ردهبندی سلسلهمراتبی را معرفی کرد (گونه، جنس، خانواده، راسته، رده، شاخه، پادشاهی)؛ اما این طبقهبندی بر اساس روابط تکاملی نبود.
۲. داروین و درخت حیات
نقطه عطف در سال ۱۸۵۹ با انتشار کتاب خاستگاه گونهها توسط چارلز داروین رقم خورد. داروین بیان کرد:
-
همه گونهها از طریق نیاکان مشترک با هم مرتبطاند
-
تکامل از طریق انتخاب طبیعی رخ میدهد
-
تاریخچه حیات را میتوان بهصورت یک "درخت حیات" ترسیم کرد که شاخههای آن نمایانگر دودمانها هستند
نقلقول معروف از داروین:
"ارتباط بین موجوداتی که به یک رده تعلق دارند، گاه به شکل درختی بزرگ نمایش داده شده است."
۳. ظهور کلادیستیک (Cladistics) در قرن ۲۰
در دهه ۱۹۵۰ تا ۶۰، علم کلادیستیک با تلاشهای ویلی هنینگ (Willi Hennig) ظهور کرد:
-
مفهوم کلاد (Clade) را معرفی کرد: گروهی شامل یک نیا و همه نوادگانش
-
تمرکز را از شباهت کلی به سمت ویژگیهای اشتراکی اشتقاقی (Synapomorphies) سوق داد
-
پایههای سیستماتیک فیلوژنتیک مدرن را بنیان گذاشت
۴. فیلوژنی مولکولی (اواخر قرن ۲۰)
با کشف DNA و پیشرفتهای زیستشناسی مولکولی، دانشمندان شروع به استفاده از توالیهای ژنتیکی (DNA، RNA، پروتئینها) برای بازسازی روابط تکاملی کردند.
-
کارل ووز (Carl Woese) در سال ۱۹۷۷ با بررسی rRNA توانست موجودات زنده را به سه قلمرو تقسیم کند: باکتریها، آرکیها و یوکاریوتها.
-
دادههای مولکولی، ابزاری عینیتر و کمیتر برای ترسیم درختهای فیلوژنتیک فراهم کردند.
۵. فیلوژنومیکس و دادههای کلان (قرن ۲۱)
انفجار در تعیین توالی ژنوم و توسعه ابزارهای محاسباتی، باعث ایجاد شاخهای به نام فیلوژنومیکس (Phylogenomics) شد:
-
استفاده از کل ژنومها یا هزاران ژن برای بررسی روابط تکاملی
-
کاربرد یادگیری ماشین و هوش مصنوعی در تحلیل درختهای پیچیده
-
پروژههایی مانند درخت باز زندگی (Open Tree of Life) و پروژه وب درخت زندگی (Tree of Life Web Project) در حال ساخت درختی جامع از تمام حیات شناختهشده هستند
چرا فیلوژنی مهم است؟
درک فیلوژنی برای تقریباً تمام شاخههای زیستشناسی حیاتی است. برخی از دلایل اهمیت آن:
۱. کشف تاریخچه تکاملی
فیلوژنی نشان میدهد که حیات چگونه در طول میلیاردها سال تنوع یافته و گونههای امروزی چگونه از نیاکان باستانی به وجود آمدهاند.
۲. کمک به ردهبندی علمی
ردهبندیهای امروزی بهطور فزایندهای بر شواهد فیلوژنتیک استوارند. مثلاً:
-
امروزه پرندگان در درون گروه دایناسورها (Theropoda) طبقهبندی میشوند، زیرا از نظر تکاملی از آنها منشأ گرفتهاند.
۳. داروسازی و پزشکی
درختهای فیلوژنتیک برای:
-
ردیابی شیوع بیماریها (مانند انواع کروناویروس)
-
شناسایی اهداف دارویی با بررسی ژنهای محافظتشده در طی تکامل
-
درک تکامل پاتوژنها و مقاومت آنتیبیوتیکی کاربرد دارند
۴. حفاظت از تنوع زیستی
فیلوژنی به شناسایی گونههای منحصر بهفرد از نظر تکاملی کمک میکند تا اولویت بیشتری برای حفاظت داشته باشند.
۵. بومشناسی و رفتارشناسی
گونههای نزدیک از نظر تکاملی، معمولاً نقشهای زیستمحیطی یا الگوهای رفتاری مشابهی دارند. درختهای فیلوژنتیک به بررسی فرضیههای تکاملی در مورد ویژگیهای رفتاری و بومشناختی کمک میکنند.
چگونه فیلوژنی را نمایش میدهند؟
درختهای فیلوژنتیک
نمودارهای شاخهای که روابط بین تاکسا (گونهها یا گروهها) را نمایش میدهند. اجزای اصلی:
-
ریشه (Root): نیاکان مشترک همه تاکسا
-
شاخهها (Branches): دودمانها در طول زمان
-
گرهها (Nodes): نقاط واگرایی؛ نماد نیاکان مشترک
-
برگها (Tips): گونهها یا تاکساهای زنده امروزی
انواع مختلف درختها:
-
کلادوگرام (Cladogram): فقط ترتیب انشعاب را نشان میدهد (بدون مقیاس زمانی)
-
فیلوگرام (Phylogram): طول شاخهها متناسب با تغییرات ژنتیکی است
-
کرونوگرام (Chronogram): طول شاخهها بیانگر زمان واقعی است
مفاهیم کلیدی در فیلوژنی
-
گروه تکتبار (Monophyletic / Clade): شامل نیا و همه نوادگانش
-
گروه شبهتبار (Paraphyletic): نیا و بخشی از نوادگان
-
گروه چندتبار (Polyphyletic): فاقد نیاکان مشترک مشخص برای همه اعضا
در سیستماتیک مدرن، فقط گروههای تکتبار معتبر شناخته میشوند.
مثال واقعی: فیلوژنی انسانها
ردهبندیهای سنتی، انسانها را در خانوادهای جدا از میمونها قرار میداد. اما شواهد فیلوژنتیک نشان میدهند که:
-
انسانها حدود ۹۸.۸٪ از DNA خود را با شامپانزهها مشترک دارند
-
نیاکان مشترک انسان و شامپانزه حدود ۵ تا ۷ میلیون سال پیش میزیستهاند
-
امروزه انسانها، شامپانزهها، گوریلها و اورانگوتانها در خانواده Hominidae (کَپیهای بزرگ) جای میگیرند، که یک کلاد (گروه تکتبار) محسوب میشود
بخش ۲: مفاهیم پایه – واژگان درخت تبارزایی، ریشهگذاری و گرهها
درک علم تبارزایی نیازمند آشنایی با اصطلاحات پایه و ساختار درختهای تبارزایی است؛ این درختها نمودارهایی هستند که روابط تکاملی بین گروهی از موجودات زنده یا ژنها را نشان میدهند. این بخش به بررسی اجزای بنیادی درختهای تبارزایی میپردازد، از جمله انواع درختها، نحوهٔ ریشهگذاری آنها، و معانی زیستی عناصر مختلفی مانند گرهها، شاخهها، دودمانها (clades) و سایر موارد.
۲.۱ درخت تبارزایی چیست؟
درخت تبارزایی (که در برخی زمینهها به آن درخت تکاملی یا کلادوگرام نیز گفته میشود)، نموداری است که روابط تکاملی بین گروهی از موجودات یا ژنها را نشان میدهد. هر نقطهٔ انشعاب در درخت، بیانگر جد مشترک است و الگوی شاخهزنی، فرضیهای دربارهٔ ترتیب و زمان رخ دادن جداییها را نشان میدهد.
به یاد داشته باشید:
درخت تبارزایی یک فرضیه است، نه یک حقیقت قطعی. این درختها بر پایهٔ دادههای موجود – چه دادههای مورفولوژیکی (ریختشناسی) یا دادههای مولکولی – ساخته میشوند و ممکن است با ظهور شواهد جدید تغییر کنند.
۲.۲ واژگان درخت تبارزایی
برای فهم و تفسیر درست درختهای تبارزایی، لازم است با واژگان کلیدی آن آشنا باشید:
-
تاکسا (Taxa) (مفرد: Taxon): موجوداتی (گونهها، جمعیتها، ژنها و...) که در نوکهای درخت (برگها) نمایش داده میشوند.
-
گره (Node): نقطهای روی درخت که در آن یک تبار به دو یا چند شاخه تقسیم میشود. گرهها شامل دو نوع هستند:
-
گرههای داخلی (Internal nodes): نشاندهندهٔ اجداد فرضی مشترک هستند.
-
گرههای انتهایی (Terminal nodes) یا برگها: گونههای فعلی یا منقرضشدهاند (تاکساهای مشاهدهشده).
-
-
شاخه (Branch یا Edge): خطی که گرهها را به هم متصل میکند. شاخه نمایانگر تبار تکاملی بین دو رویداد جدایی است و ممکن است طول آن متناسب با زمان تکاملی یا تغییرات ژنتیکی باشد.
-
دودمان (Clade): گروهی تکنیا (Monophyletic)، شامل یک جد مشترک و تمامی نوادگان آن.
-
ریشه (Root): پایینترین گره در درخت که نشاندهندهٔ نزدیکترین جد مشترک تمام تاکساها در درخت است. درختها میتوانند ریشهدار یا بدون ریشه (Unrooted) باشند.
-
گروههای خواهری (Sister groups): دو دودمان یا تاکسایی که نزدیکترین خویشاوندان یکدیگر هستند؛ یعنی جد مشترک مستقیمی دارند.
-
چندشاخهای (Polytomy): گرهای با بیش از دو شاخه؛ نشاندهندهٔ ابهام یا نبود قطعیت در روابط تکاملی است.
-
گروه بیرونی (Outgroup): تاکسا یا گروهی از تاکساها که مشخصاً پیش از گروه مورد نظر (Ingroup) از تبار اصلی جدا شدهاند و برای ریشهگذاری درخت استفاده میشوند.
۲.۳ انواع درختهای تبارزایی
درختهای تبارزایی را میتوان به شکلهای مختلفی نمایش داد که هرکدام مفاهیم خاصی دارند:
-
کلادوگرامها (Cladograms):
-
فقط ترتیب شاخهزنی را نشان میدهند.
-
طول شاخهها بیانگر زمان یا تغییرات نیست.
-
برای نمایش روابط بدون فرض دربارهٔ نرخ تغییرات مناسب هستند.
-
-
فایلوگرامها (Phylograms):
-
طول شاخهها متناسب با میزان تغییرات تکاملی (مثلاً تعداد جهشهای نوکلئوتیدی) است.
-
معمولاً بر پایهٔ دادههای مولکولی ساخته میشوند.
-
-
کرونوگرامها (Chronograms):
-
درختهایی هستند که طول شاخهها با زمان واقعی مقیاسبندی شدهاند (با استفاده از فسیلها یا ساعت مولکولی).
-
برای درک زمان جداییها مفید هستند.
-
۲.۴ ریشهگذاری درخت تبارزایی
ریشهگذاری درخت تبارزایی جهت تکامل را مشخص میکند و به تعیین نقطهٔ نخستین انشعاب و جد مشترک بین تاکساهای موجود در درخت کمک مینماید. انتخاب روش مناسب برای ریشهگذاری تأثیر بسزایی در تفسیر روابط تکاملی دارد.
روشهای متداول ریشهگذاری عبارتاند از:
-
استفاده از گروه بیرونی (Outgroup):
رایجترین و معتبرترین روش برای ریشهگذاری، بهرهگیری از یک یا چند تاکسای بیرونی است که مشخصاً پیش از سایر تاکسا از تبار مشترک جدا شدهاند. استفاده از گروه بیرونی، جهت تغییرات تکاملی را مشخص کرده و امکان تعیین جد مشترک گروه هدف را فراهم میسازد. -
ریشهگذاری با نقطهٔ میانی (Midpoint Rooting):
در این روش، ریشه در نقطهٔ میانی طولانیترین مسیر بین دو تاکسا قرار میگیرد. این رویکرد بر پایهٔ فرض وجود نرخ تکاملی یکنواخت در طول شاخهها (ساعت مولکولی) استوار است. با این حال، در صورت نابرابری نرخ تغییرات در شاخهها، دقت آن کاهش مییابد. -
درختهای بدون ریشه (Unrooted Trees):
این نوع درختها تنها نشاندهندهٔ روابط نسبی میان تاکسا هستند، بدون آنکه جهت تکامل را مشخص کنند. در مواردی که اطلاعات لازم برای تعیین ریشه در دسترس نیست یا الگوریتمهای مورد استفاده جهتدار نیستند (مانند روش Neighbor-Joining)، از درختهای بدون ریشه استفاده میشود.
۲.۵ گرهها و تفسیر زیستی آنها
گرهها کلید درک روابط تبارزایی هستند و میتوانند معانی مختلفی داشته باشند:
-
رویدادهای گونهزایی (Speciation): اغلب، گره داخلی نشاندهندهٔ تجزیهٔ یک تبار به دو گونهٔ جدید است.
-
رویدادهای دوبرابر شدن ژن (Gene Duplication): در درختهای ژنی، گره ممکن است نشاندهندهٔ تکثیر ژن باشد، نه گونهزایی.
-
انتقال افقی ژن یا دورگهسازی (Hybridization): در برخی موارد، گره حاصل از وراثت غیرعمودی است که به تکامل شبکهای منجر میشود. در چنین شرایطی از درختهای شبکهای (networks) استفاده میشود.
۲.۶ گروههای تکنیا، پارانیا و چندنیا
درک نوع گروهبندیها در درخت برای تفسیر درست تاکسونومی ضروری است:
-
گروه تکنیا (Monophyletic / Clade): شامل جد مشترک و تمام نوادگانش است.
-
تنها نوع گروهی که در کلادیستیک معتبر محسوب میشود.
-
-
گروه پارانیا (Paraphyletic): شامل جد مشترک، اما نه همهٔ نوادگان.
-
مثلاً اگر پرندگان را از «خزندگان» حذف کنیم، خزندگان گروهی پارانیا خواهند بود.
-
-
گروه چندنیا (Polyphyletic): شامل موجوداتی که جد مشترک نزدیکی ندارند.
-
معمولاً بهدلیل شباهتهای همگرا (Convergent traits) گروهبندی شدهاند.
-
۲.۷ همولوژی در برابر هموپلازی
در ساخت و تفسیر درختها، باید تفاوت شباهت ناشی از اجداد مشترک با شباهتهای ظاهریِ ناشی از عوامل دیگر را تشخیص داد:
-
همولوژی (Homology): ویژگیای که از جد مشترک به ارث رسیده است.
-
مثال: اندامهای حرکتی در مهرهداران.
-
-
هموپلازی (Homoplasy): ویژگیای که مشابه بهنظر میرسد اما منشأ مشترک ندارد؛ نتیجهٔ تکامل همگرا یا بازگشت تکاملی است.
-
مثال: بال در خفاش و پرنده.
-
-
هموپلازی میتواند تفسیر درخت را گمراه کند و نیازمند استفاده از مدلهای پیچیده برای اصلاح آن است.
۲.۸ اعتبار شاخهها و میزان اطمینان
از آنجا که درختها بر پایهٔ روشهای استنباطی ساخته میشوند، باید آنها را با دید احتمالی تفسیر کرد. برای ارزیابی اعتبار شاخهها از روشهایی استفاده میشود:
-
مقادیر Bootstrap: روش نمونهگیری مجدد برای ارزیابی پشتیبانی؛
-
مقادیر بیشتر یا برابر ۷۰٪ قابلاعتماد در نظر گرفته میشوند.
-
-
احتمالهای پسین (Posterior Probabilities): در روشهای بیزی،
-
مقادیر نزدیک به ۱ نشانهٔ حمایت قوی هستند.
-
-
امتیاز احتمال (Likelihood Scores): در روش حداکثر درستنمایی،
-
بهترین درخت، درختی است که احتمال تولید دادههای مشاهدهشده در آن بیشینه باشد.
-
۲.۹ توپولوژی درخت در برابر طول شاخهها
-
توپولوژی (Topology): الگوی روابط – یعنی اینکه چه کسی با چه کسی خویشاوندتر است.
-
طول شاخهها: نشاندهندهٔ مقدار تغییر تکاملی یا زمان است – بسته به نوع درخت.
دو درخت میتوانند توپولوژی یکسان ولی طول شاخههای متفاوت داشته باشند، بسته به دادهها یا مدل استفادهشده.
۲.۱۰ تفسیر نمودارهای درختی
نکتهٔ بسیار مهم در خواندن درختها این است که:
-
جهتگیری نمودار (چپ، راست، بالا یا پایین بودن تاکسا) هیچ تأثیری بر روابط ندارد.
-
چرخاندن گرهها حول یک نقطه، معنی درخت را تغییر نمیدهد، تا زمانی که ترتیب شاخهزنی حفظ شود.
-
نزدیکی بصری تاکسا نشاندهندهٔ خویشاوندی نزدیکتر نیست، مگر آنکه آنها جد مشترک جدیدتری داشته باشند.
بخش ۳: انواع شواهد تبارزایی – ریختشناسی، مولکولی و ژنومی
ساخت درختهای تبارزایی به در دسترس بودن دادههایی قابلاعتماد که بازتابدهندهٔ روابط تکاملی باشند، وابسته است. در این بخش، سه دستهٔ اصلی از شواهد که برای استنتاج این روابط بهکار میروند بررسی میشوند:
-
شواهد ریختشناسی (Morphological Evidence)
-
شواهد مولکولی (Molecular Evidence)
-
شواهد ژنومی و فایلوژنومیک (Genomic and Phylogenomic Evidence)
هر نوع داده مزایا، محدودیتها و کاربردهای خاص خود را دارد. شناخت ویژگیهای آنها برای انتخاب منابع دادهٔ مناسب و طراحی تحلیلهای تبارزایی قوی ضروری است.
۳.۱ شواهد ریختشناسی
تعریف و مثالها
دادههای ریختشناسی به ویژگیهای فیزیکی قابلمشاهدهٔ موجودات زنده اشاره دارند، مانند:
-
ویژگیهای ظاهری بیرونی (مثلاً شکل اندام، تقسیمبندی بدن)
-
آناتومی داخلی (مانند ساختار استخوانها، سیستمهای اندامی)
-
الگوهای رشد و نمو (مثلاً جنینشناسی)
-
رفتارها (مانند نمایشهای جفتگیری)
این ویژگیها بهصورت ویژگیهایی با حالتهای متفاوت (مانند "بال دارد" در برابر "بال ندارد") کدگذاری میشوند و در تحلیلهای کلادیستی برای استنتاج روابط بر پایهٔ صفات اشتراکی اشتقاقیافته (Synapomorphies) استفاده میشوند.
کاربردها
-
گونههای فسیلی (که معمولاً DNA آنها در دسترس نیست)
-
آناتومی تطبیقی در سیستماتیک
-
حل روابط تکاملی عمیق میان گروههای اصلی
مزایا
-
جهانشمول بودن: ویژگیهای ریختشناسی برای همه موجودات شناختهشده، زنده و منقرضشده، در دسترس هستند.
-
امکان درج فسیلها: این دادهها اجازه میدهند که گونههای منقرضشده در درختهای تبارزایی گنجانده شوند.
-
ارتباط مستقیم با فنوتیپ: صفات ریختشناسی مستقیماً با سازگاری بومشناختی و عملکردی مرتبطاند.
محدودیتها
-
تکامل همگرا (Convergent Evolution) شایع است (مثلاً وجود بال در پرندگان، خفاشها و حشرات).
-
صفات ریختشناسی ممکن است کمتعداد یا مبهم باشند.
-
کدگذاری صفات میتواند ذهنی باشد.
-
تشخیص همولوژی ممکن است دشوار باشد.
-
دقت کم برای تاکساهای بسیار نزدیکبههم.
مثال موردی
در قرن نوزدهم و اوایل قرن بیستم، زیستشناسان برای ردهبندی مهرهداران از صفات ریختشناسی استفاده کردند. برای مثال، داروین و هاکسلی با بهرهگیری از دادههای اسکلتی و جنینی، پیش از شواهد مولکولی، نزدیکی پرندگان و خزندگان را مطرح کردند.
۳.۲ شواهد مولکولی
با پیشرفت زیستشناسی مولکولی، توالیهای DNA و پروتئینها به ابزارهای قدرتمند برای استنباط تبارزایی تبدیل شدند.
انواع دادههای مولکولی
-
توالیهای DNA:
-
DNA میتوکندریایی (mtDNA)
-
ژنهای هستهای
-
ژنهای RNA ریبوزومی (مانند 16S، 18S)
-
-
توالیهای RNA
-
توالیهای پروتئین
-
میکروستالیتها و SNPها (جهشهای تکنوکلئوتیدی)
در تبارزایی مولکولی، توالیها از تاکساهای مختلف همتراز (Align) شده و برای یافتن شباهتها و تفاوتها مقایسه میشوند. این تفاوتها (مانند جانشینیها، درج یا حذفها) بازتابی از واگرایی تکاملی هستند.
کاربردها
-
استنتاج روابط میان گونههای نزدیکبههم
-
بازسازی شاخههای عمیق درخت زندگی
-
تاریخگذاری رویدادهای جدایی با استفاده از ساعت مولکولی
-
بررسی انتقال افقی ژن، دورگهسازی و درونریزی (Introgression)
مزایا
-
وضوح بالا: امکان مقایسهٔ دقیق حتی بین موجودات بسیار شبیه به هم.
-
حجم داده زیاد: امکان استفاده از کل ژنومها یا رونویسیها (ترنسکریپتومها).
-
کمی و آماری: تفاوتها قابل اندازهگیری و تحلیل آماری هستند.
-
قابل خودکارسازی و بازتولید: تراز کردن، برازش مدل و ساخت درخت میتواند توسط رایانهها استانداردسازی شود.
محدودیتها
-
نیاز به DNA با کیفیت بالا و قابل استخراج (که در بسیاری از فسیلها وجود ندارد).
-
هموپلازی ناشی از جانشینیهای متعدد در یک جایگاه (بهویژه در نواحی با تکامل سریع).
-
ناسازگاری میان درخت ژن و درخت گونه (Incomplete Lineage Sorting)
-
نیازمند ابزارهای بیوانفورماتیکی و مدلهای آماری پیشرفته
-
انتقال افقی ژن در میکروارگانیسمها میتواند روابط را مبهم کند.
مثال موردی
در سال ۱۹۷۷، کارل ووز (Carl Woese) با استفاده از توالیهای rRNA 16S نشان داد که آرکیها (Archaea) حوزهای مجزا از زندگی هستند، جدا از باکتریها و یوکاریوتها. این کشف، فهم ما از درخت زندگی را دگرگون کرد.
۳.۳ شواهد ژنومی و فایلوژنومیک
فایلوژنومیک چیست؟
فایلوژنومیک تحلیل کل ژنومها یا بخشهای بزرگی از آنها برای استنباط روابط تکاملی است. برخلاف تبارزایی مولکولی سنتی که معمولاً فقط از چند ژن استفاده میکند، فایلوژنومیک هزاران جایگاه ژنومی را بررسی میکند.
روشهای فایلوژنومیک میتوانند شامل موارد زیر باشند:
-
توالییابی کل ژنوم
-
ترنسکریپتومیکس (RNA-seq)
-
کپچر اگزوم (Exome Capture)
-
عناصر بسیار محافظتشده (Ultra-Conserved Elements)
-
ارتولوگهای تککپی (Single-Copy Orthologs)
کاربردها
-
ساخت درختهای با وضوح بسیار بالا در تمامی مقیاسهای تاکسونومیک
-
حل روابط دشوار یا بحثبرانگیز
-
بررسی تکامل سازشی و گسترش خانوادههای ژنی
-
تشخیص گونهها، بهویژه در گونههای پنهان (Cryptic Species)
-
مطالعه دورگهسازی و درونریزی ژنومی
مزایا
-
حجم عظیم دادهها دقت و استحکام تحلیل را افزایش میدهد.
-
کاهش سوگیریهای ژنمحور (مانند اشباع یا تغییر نرخ تکاملی).
-
امکان برآورد درخت گونهها بهجای فقط درخت ژنها.
-
مناسب برای جداییهای کمعمق و پرعمق.
محدودیتها
-
نیازمند محاسبات سنگین و رایانههای قدرتمند.
-
کیفیت داده بسیار مهم است؛ آلودگی یا مونتاژ اشتباه میتواند نتایج را منحرف کند.
-
ناسازگاری درختهای ژنی به دلیل نوترکیبی، تکثیر یا ILS.
-
هزینه بالا (اگرچه با فناوریهای نوین رو به کاهش است).
مثال موردی
در سال ۲۰۱۵، یک مطالعهٔ فایلوژنومیک مهم توسط Jarvis و همکاران با استفاده از دادههای ژنوم کامل ۴۸ گونه پرنده انجام شد و درخت تبارزایی پرندگان را با دقت بالا بازسازی کرد، و به بحثهای طولانی درباره جایگاه گروههایی مثل پرندگان آوازخوان، پرندگان شکاری و پرندگان آبی پایان داد.
۳.۴ مقایسه انواع شواهد
|
ویژگی |
ریختشناسی |
مولکولی |
ژنومی |
|
منبع داده |
صفات فیزیکی |
توالی ژن یا پروتئین |
دادههای ژنوم کامل |
|
دسترسپذیری |
همهٔ تاکسا |
نیازمند DNA زنده یا خوب حفظشده |
محدود به تاکساهایی با ژنوم توالییابیشده |
|
وضوح (Resolution) |
کم تا متوسط |
متوسط تا زیاد |
بسیار بالا |
|
درج فسیل |
بله |
بهندرت (فقط DNA باستانی) |
نه (مگر فسیلهای تازه) |
|
ریسک همگرایی |
زیاد |
متوسط (بسته به نشانگر) |
متغیر |
|
هزینه |
کم |
متوسط |
بالا |
|
نیاز محاسباتی |
کم |
متوسط |
بسیار بالا |
۳.۵ ادغام انواع دادهها
تبارزایی مدرن اغلب از ترکیب چند نوع داده بهره میبرد – فرآیندی که به آن تحلیل شواهد کل (Total Evidence Analysis) یا تبارزایی یکپارچه گفته میشود.
برای مثال:
-
دادههای ریختشناسی و مولکولی را میتوان در چارچوبهای بیزی (مانند MrBayes) ترکیب کرد تا گونههای فسیلی و زنده را همزمان در درخت گنجاند.
-
دادههای ژنومی را میتوان فیلتر کرد تا ژنهای غیرارتولوگ حذف شوند، و دقت افزایش یابد.
-
دادههای ترنسکریپتومی میتوانند در مواردی که DNA موجود نیست، اما RNA قابل توالییابی است، شکافها را پر کنند.
این رویکردهای تلفیقی به اعتبار نتایج، پشتیبانی درخت و کشف الگوهای پنهان در تکامل کمک شایانی میکنند.
نتیجهگیری بخش ۳
نوع دادههایی که در استنباط تبارزایی بهکار میروند، شکل نهایی درخت را تعیین میکنند.
-
دادههای ریختشناسی امکان درج فسیلها و کاربرد گسترده دارند اما در برابر همگرایی آسیبپذیرند و میتوانند مبهم باشند.
-
دادههای مولکولی وضوح بالا و پایهٔ آماری قوی فراهم میکنند.
-
رویکردهای ژنومی عمق و دقت بیسابقهای دارند، اگرچه پیچیدگی زیادی دارند.
تحلیل کامل تبارزایی اغلب نیاز به ترکیب استراتژیک این منابع دارد، بسته به پرسش پژوهشی، گروه تاکسونومیک مورد بررسی، و منابع در دسترس.
در گام بعدی، هنگام ورود به روشهای ساخت درخت، شناخت ماهیت دادههای ورودی برای انتخاب روش تحلیلی مناسب، ضروری است.
بخش ۴: روشهای ساخت درخت تبارزایی – مبتنی بر فاصله، ویژگی و بیزین
پس از جمعآوری و همترازسازی صحیح دادههای تبارزایی (ریختشناسی، مولکولی یا ژنومی)، گام بعدی و حیاتی، ساخت درخت تبارزایی است – یعنی نموداری که فرضیهای دربارهٔ روابط تکاملی بین تاکسا (گروههای زیستی) ارائه میدهد.
فرایند ساخت درخت از میان چندین روش آماری یا الگوریتمی انجام میشود که هرکدام فرضیات، مزایا و محدودیتهای خاص خود را دارند.
در این بخش، چهار دستهٔ اصلی از روشها بررسی میشوند:
-
روشهای مبتنی بر فاصله (Distance-Based Methods)
-
روشهای مبتنی بر ویژگی (Character-Based Methods)
-
استنتاج بیزین (Bayesian Inference)
-
مقایسه و ملاحظات مربوط به انتخاب روش
۴.۱ روشهای مبتنی بر فاصله (Distance-Based)
این روشها ابتدا تفاوتهای توالی را به صورت ماتریسی از فواصل زوجی بین تاکسا تبدیل میکنند و سپس با استفاده از این ماتریس، درخت را میسازند. برخلاف روشهای ویژگیمحور، این روشها ویژگیهای منفرد را مستقیماً بررسی نمیکنند، بلکه شباهت کلی را خلاصه میکنند.
الف) روش UPGMA (Unweighted Pair Group Method with Arithmetic Mean)
-
فرض اصلی: ثابت بودن نرخ تکامل (فرض ساعت مولکولی)
-
نحوه عملکرد: تاکسا را بر اساس میانگین حداقل فواصل زوجی خوشهبندی میکند
-
خروجی: درختی ریشهدار با مسیرهای مساوی از ریشه تا برگها
مزایا:
-
ساده و سریع
-
مناسب برای تحلیلهای مقدماتی
محدودیتها:
-
وابسته به فرض سخت ساعت مولکولی – اگر این فرض نقض شود، درخت اشتباه خواهد بود
-
بیش از حد سادهسازی میکند و برای دادههای پیچیده مناسب نیست
ب) روش Neighbor-Joining (NJ)
-
فرض ساعت مولکولی ندارد
-
هدف: کمینهسازی طول کلی شاخهها
-
اغلب در تحلیل دادههای توالی DNA استفاده میشود
مزایا:
-
کارآمد و سریع، حتی با مجموعهدادههای بزرگ
-
خروجی: درخت بدون ریشه (Unrooted)
-
مناسب برای تحلیلهای اولیه یا bootstrap
محدودیتها:
-
بر پایهٔ مدلهای تکاملی صریح نیست
-
حساس به خطاهای محاسبه فاصله
مثال کاربردی:
NJ یکی از روشهای رایج در نرمافزار MEGA برای ساخت درخت از دادههایی مانند توالی ژن 16S rRNA است.
۴.۲ روشهای مبتنی بر ویژگی (Character-Based)
برخلاف روشهای فاصلهای، این روشها هر ویژگی (مثلاً جایگاه نوکلئوتیدی یا صفت ریختی) را بهطور جداگانه بررسی میکنند و مسیرهای تکاملی را در نظر میگیرند.
الف) حداکثر صرفهجویی (Maximum Parsimony - MP)
-
اصل: بهترین درخت درختی است که کمترین تعداد تغییرات تکاملی را نیاز دارد
-
برگرفته از اصل اُکام (Occam’s Razor): سادهترین توضیح بهترین است
نحوه عملکرد:
-
هر سایت/ویژگی به عنوان یک کاراکتر در نظر گرفته میشود
-
تمام درختهای ممکن را بررسی کرده و آنکه مجموع تغییراتش حداقل است انتخاب میشود
مزایا:
-
ساده و شهودی
-
قابل استفاده برای دادههای ریختشناسی و مولکولی
-
نیازی به مدل تکاملی صریح ندارد
محدودیتها:
-
حساس به هموپلازی (تکامل همگرا، برگشتهای تکاملی)
-
فرض میکند همه تغییرات وزن و احتمال مساوی دارند
-
زمانبر و سنگین محاسباتی برای دادههای بزرگ
مثال کاربردی:
MP در دیرینشناسی بسیار مفید است؛ جایی که ویژگیهای فسیلی مانند استخوانها در گونههای منقرض بررسی میشوند.
ب) حداکثر درستنمایی (Maximum Likelihood - ML)
-
رویکرد آماری: احتمال (Likelihood) مشاهدهٔ دادهها را با توجه به یک درخت و مدل تکاملی خاص محاسبه میکند
-
بهترین درخت، درختی است که بیشترین احتمال را دارد
اجزای کلیدی:
-
مدلهای جانشینی (Substitution Models) مانند:
-
Jukes-Cantor، Kimura، GTR
-
-
ناهمگنی نرخها در مکانهای مختلف (مثلاً توزیع گاما)
مزایا:
-
از لحاظ آماری قدرتمند
-
توانایی استفاده از مدلهای پیچیدهٔ تکامل
-
میتوان با آن فرضیهها را با آزمون نسبت درستنمایی (LRT) آزمود
محدودیتها:
-
محاسبات سنگین
-
نیاز به انتخاب مدل مناسب (مدل ضعیف = درخت ضعیف)
-
کندتر از NJ یا MP بهویژه در دادههای بزرگ
مثال کاربردی:
روش ML در نرمافزارهای قدرتمندی مانند RAxML، PhyML، IQ-TREE برای بازسازی درختهای تبارزایی استفاده میشود.
۴.۳ استنتاج بیزین (Bayesian Inference)
در تبارزایی بیزین، از قضیه بیز برای تخمین احتمال پسین درختها با توجه به دادهها استفاده میشود.
اصل بنیادی بیز:
P(Tree∣Data)=P(Data∣Tree)×P(Tree)P(Data)\mathbf{P(Tree | Data)} = \frac{\mathbf{P(Data | Tree)} \times \mathbf{P(Tree)}}{\mathbf{P(Data)}}P(Tree∣Data)=P(Data)P(Data∣Tree)×P(Tree)
-
P(Tree | Data): احتمال پسین (درخت مورد نظر ما)
-
P(Data | Tree): درستنمایی (مشابه ML)
-
P(Tree): احتمال پیشین درخت (Prior)
-
P(Data): درستنمایی نهایی داده (ثابت)
پیادهسازی در: MrBayes، BEAST، RevBayes
ویژگیهای کلیدی:
-
به جای یک درخت، مجموعهای از درختهای محتمل تولید میکند
-
از روش زنجیره مارکف مونتکارلو (MCMC) برای نمونهبرداری از فضای درختها استفاده میکند
-
میتواند دانش قبلی (مثلاً سن فسیل، دودمانهای شناختهشده) را در تحلیل وارد کند
مزایا:
-
ارائه مقادیر اعتماد (احتمال پسین) برای شاخهها
-
مدلسازی انعطافپذیرتر (مانند ساعتهای غیرسختگیرانه، مدلهای تولد-مرگ)
-
امکان تخمین همزمان زمان جدایی و توپولوژی درخت
محدودیتها:
-
بسیار پرهزینه از نظر محاسباتی
-
نیاز به انتخاب دقیق پیشینها (priors)
-
تفسیر مقادیر احتمال پسین میتواند در صورت انتخاب مدل غلط گمراهکننده باشد
مثال کاربردی:
در همهگیریشناسی، روش بیزین (مثلاً با BEAST) برای ردیابی دودمانهای SARS-CoV-2 و برآورد نرخ جهش و زمان انتقال استفاده شده است.
۴.۴ انتخاب مدل در ML و بیزین
انتخاب مدل تکاملی مناسب اهمیت زیادی دارد.
مدلهای رایج برای DNA:
-
JC69 (Jukes-Cantor): همه جانشینیها احتمال برابر دارند
-
K80 (Kimura): تمایز بین جانشینیها و جابهجاییها
-
HKY، GTR: نرخهای متفاوت جانشینی برای نوکلئوتیدها
ابزارهای انتخاب مدل:
-
jModelTest
-
ModelFinder
-
PartitionFinder
مدل مناسب باید تعادل بین برازش و پیچیدگی را حفظ کند، معمولاً با معیارهایی مانند:
-
Akaike Information Criterion (AIC)
-
Bayesian Information Criterion (BIC)
-
Likelihood Ratio Tests (LRTs)
۴.۵ Bootstrap و مقادیر پشتیبانی
مهم نیست که از چه روشی استفاده میکنید؛ بررسی آماریِ میزان اعتماد به شاخهها ضروری است.
تحلیل Bootstrap (ویژه روشهای ML و NJ):
-
دادهها را چندین بار بازنمونهگیری کرده و درخت را میسازد
-
تعداد تکرار: معمولاً ۱۰۰ تا ۱۰۰۰
-
شاخههایی با پشتیبانی بالای ۷۰٪ قابلاعتماد در نظر گرفته میشوند
احتمالات پسین (Posterior Probabilities در روش بیزین):
-
نشاندهندهٔ درصد درختهایی هستند که یک شاخه در آنها ظاهر شده است
-
مقادیر بالای ۰.۹۵ نشانهٔ حمایت قوی هستند
۴.۶ جدول خلاصه روشها
|
روش |
نوع |
نیاز به مدل؟ |
سرعت |
نوع داده |
ارزش پشتیبانی |
|
UPGMA |
فاصلهای |
خیر |
بسیار سریع |
مولکولی |
ندارد |
|
NJ |
فاصلهای |
خیر |
سریع |
مولکولی |
Bootstrap |
|
MP |
ویژگیمحور |
خیر |
متوسط |
ریختی و مولکولی |
Bootstrap |
|
ML |
ویژگیمحور |
بله |
کند |
مولکولی |
Bootstrap |
|
Bayesian |
ویژگیمحور |
بله |
بسیار کند |
مولکولی |
Posterior Probability |
۴.۷ انتخاب روش مناسب
|
سناریو |
روش پیشنهادی |
|
درخت اکتشافی با داده DNA |
Neighbor-Joining (NJ) |
|
ویژگیهای فسیلی یا آناتومیک |
Maximum Parsimony (MP) |
|
استنتاج دقیق بر پایه مدل |
Maximum Likelihood (ML) |
|
درخت با برآورد زمان جدایی |
Bayesian (مثلاً BEAST) |
|
دادههای ژنومی گسترده |
ML (مانند IQ-TREE) یا Bayesian با ساعت غیرسختگیرانه |
نتیجهگیری بخش ۴
ساخت درخت تبارزایی ترکیبی از علم و هنر است. انتخاب روش مناسب بستگی دارد به:
-
نوع دادههای شما
-
اهداف پژوهش
-
منابع محاسباتی
-
فرضیات تکاملی
با پیشرفت فناوریهای توالییابی و مدلسازی، توانایی ما برای ساخت درختهای تبارزایی دقیق، جامع و با پشتیبانی آماری بالا نیز بیشتر شده است.
شناخت مزایا و معایب هر روش تضمین میکند که درخت نهایی نهتنها از نظر محاسباتی صحیح، بلکه از نظر زیستی نیز معنادار باشد.
بخش ۵: ساعت مولکولی و تخمین زمان جدایی گونهها
یکی از اهداف اصلی در مطالعات فیلوژنتیک، فقط فهمیدن این نیست که «چه کسی با چه کسی» خویشاوند است، بلکه این است که این روابط خویشاوندی در چه زمانی در تاریخ تکاملی از هم جدا شدهاند. فرضیهی ساعت مولکولی به دانشمندان این امکان را میدهد که زمان رویدادهای تکاملی را با استفاده از دادههای ژنتیکی تخمین بزنند.
در این بخش به موضوعات زیر میپردازیم:
-
مفهوم ساعت مولکولی
-
روشهای کالیبراسیون
-
ساعتهای سختگیرانه (strict) در برابر ساعتهای منعطف (relaxed)
-
مدلهای تغییر نرخ جهش
-
درختهای زمانکالیبرهشده و نقش فسیلها
-
ابزارهای مورد استفاده در تاریخگذاری مولکولی
-
کاربردها و محدودیتها
۵.۱ فرضیه ساعت مولکولی
برای نخستین بار توسط «زوکِرکَندل» و «پالینگ» در دهه ۱۹۶۰ مطرح شد. این فرضیه بیان میکند که:
جهشهای ژنتیکی با نرخ نسبتاً ثابتی در طول زمان رخ میدهند، بنابراین میتوان از تفاوتهای ژنتیکی به عنوان شاخصی برای زمان جدایی دو دودمان استفاده کرد.
یعنی:
-
تفاوت بیشتر در توالی DNA → فاصله زمانی بیشتر از زمان جدایی
-
تفاوت کمتر → اجداد مشترک اخیرتر
این اصل، پایهی درختهای فیلوژنتیک زماندار (time-calibrated trees) است.
🔸 مثال:
اگر دو گونه در DNA میتوکندریایی خود ۵٪ تفاوت داشته باشند، و نرخ جهش برابر با ۱٪ در هر میلیون سال باشد، آنگاه این دو گونه حدود ۵ میلیون سال پیش از هم جدا شدهاند.
۵.۲ کالیبرهکردن ساعت مولکولی
برای تبدیل تفاوتهای ژنتیکی به «سال» یا «میلیون سال»، باید ساعت را با شواهد خارجی تنظیم (کالیبره) کرد.
روشهای رایج کالیبراسیون:
الف. فسیلها
-
قدیمیترین فسیل شناختهشده از یک دودمان، حداقل زمان جدایی آن را مشخص میکند.
-
مثال: اگر قدیمیترین فسیل پرنده ۱۵۰ میلیون سال قدمت داشته باشد، جدایی پرندگان باید حداقل مربوط به آن زمان باشد.
ب. شواهد زیستجغرافیایی
-
مبتنی بر رویدادهای زمینشناسی شناختهشده (مانند جدایی قارهها یا تشکیل جزایر)
-
مثال: اگر جدایی یک دودمان بعد از جدا شدن آمریکای جنوبی و آفریقا اتفاق افتاده باشد، باید قدمت آن بیش از ۱۰۰ میلیون سال باشد.
ج. DNA باستانی
-
در ویروسها یا ارگانیسمهای سریعالتکامل، نمونههای قدیمی با تاریخگذاری کربنی میتوانند مستقیماً نرخ جهش را مشخص کنند.
د. کالیبراسیون ثانویه
-
استفاده از نتایج مطالعات قبلی (کماعتبارتر بوده و تنها در صورت اجبار توصیه میشود)
۵.۳ ساعتهای سختگیرانه و منعطف
ساعت سختگیرانه (Strict Clock):
-
فرض میکند نرخ جهش در همه دودمانها یکسان است.
-
ساده و محاسباتی سریع
-
در اغلب دادههای واقعی، این فرض صادق نیست
ساعت منعطف (Relaxed Clock):
-
اجازه میدهد نرخ جهش بین دودمانها متفاوت باشد
-
برای ارگانیسمهای پیچیده و دورههای زمانی طولانی واقعیتر است
-
در نرمافزارهای مدرن مانند BEAST و MrBayes پیادهسازی شده
انواع ساعتهای منعطف:
-
Lognormal نامرتبط (Uncorrelated Lognormal): نرخ جهش بهصورت تصادفی از توزیع لگنرمال انتخاب میشود
-
خودهمبسته (Autocorrelated): نرخ جهش در شاخههای مجاور مشابه است (یعنی نرخها به تدریج تغییر میکنند)
۵.۴ مدلهای تغییر نرخ جهش
نرخ جهش ممکن است به دلایل زیر متفاوت باشد:
-
زمان نسل: گونههایی با طول عمر کوتاهتر، نسلهای بیشتری در یک بازه زمانی دارند → جهش سریعتر
-
نرخ متابولیسم: متابولیسم بالاتر → تولید رادیکالهای آزاد بیشتر → جهش بیشتر
-
مکانیسمهای ترمیم DNA: ترمیم مؤثرتر → جهش کمتر
مدلها باید موارد زیر را در نظر بگیرند:
-
تفاوت نرخ بین دودمانها
-
تفاوت نرخ بین جایگاههای ژنی
-
پارتیشنبندی ژنوم (نرخهای جداگانه برای بخشهای مختلف ژنوم)
۵.۵ ساخت درختهای زماندار (Time-Calibrated)
مراحل کار:
-
انتخاب روش ساخت درخت که از تخمین زمان پشتیبانی کند (معمولاً رویکرد بیزی)
-
همتراز کردن توالیهای ژنتیکی
-
انتخاب مدلهای جایگزینی مناسب برای هر بخش
-
تعیین نوع ساعت (سختگیرانه یا منعطف)
-
افزودن محدودیتهای فسیلی یا نودی
-
اجرای تحلیل با استفاده از نمونهبرداری مارکوفی (MCMC) برای تخمین توزیع سنی گرهها
🔸 مثال:
با استفاده از نرمافزار BEAST، یک درخت زماندار از پستانداران میتواند زمانهای جدایی زیر را نشان دهد:
-
جدایی نخستسانان از جوندگان: ~۹۰ میلیون سال پیش
-
جدایی انسان از شامپانزه: ~۶ میلیون سال پیش
۵.۶ فسیلها و روش Tip-Dating
برخلاف روشهای سنتی که از فسیلها فقط برای نودها استفاده میکردند، در Tip-Dating:
-
فسیلها به عنوان نوکهای واقعی درخت در نظر گرفته میشوند
-
هر فسیل دارای تاریخ مشخص و دادههای ریختشناسی است
-
از ترکیب دادههای مورفولوژیک و مدلهای ساعت استفاده میشود
✅ این روش به تاریخگذاری شواهد کلنگر (Total-Evidence Dating) منجر میشود: ترکیب گونههای زنده و منقرضشده در یک درخت واحد
۵.۷ نرمافزارهای رایج برای تاریخگذاری مولکولی
|
نرمافزار |
نوع |
ویژگیها |
|
BEAST |
بیزی |
درخت زماندار، ساعتهای منعطف، تاریخگذاری جدایی |
|
MrBayes |
بیزی |
پشتیبانی از مدلهای ساعت، توزیع پسین (posterior) |
|
treePL |
مبتنی بر ML |
سریع برای درختهای بزرگ |
|
r8s |
نیمهپارامتریک |
قدیمیتر، هنوز مفید با فسیلها |
|
MCMCTree (PAML) |
بیزی |
سریع برای مجموعههای بزرگ، انعطاف در کالیبراسیون |
۵.۸ کاربردهای تاریخگذاری مولکولی
-
خط زمان تکامل: مثل تخمین زمان پیدایش گلها یا پرواز
-
مطالعات ماکروتکاملی: بررسی انفجارهای تکاملی مثل انفجار کامبرین
-
زیستجغرافیای تاریخی: زمانبندی جابهجایی گونهها بر اثر حرکات قارهای
-
تکامل ویروسها: ردیابی گسترش ویروسهایی مانند آنفلوآنزا، HIV و کرونا
-
حفاظت زیستی: شناسایی دودمانهای باستانی ارزشمند برای حفظ تنوع زیستی
۵.۹ محدودیتها و چالشها
-
ناقص بودن فسیلها: فقط میتوان حداقل زمان جدایی را مشخص کرد
-
تغییر نرخ جهش: حتی مدلهای منعطف هم ممکن است سادهسازی بیش از حد داشته باشند
-
عدم قطعیت در کالیبراسیون: فسیل اشتباه = زمانبندی اشتباه
-
ناهمگونی نرخها: برخی ژنها خیلی سریع یا خیلی کند تکامل مییابند
-
تخمین زمانها یک عدد قطعی نیست → همیشه با بازهی عدم قطعیت همراه است
(مثلاً «جدایی = ۴٫۲ ± ۰٫۸ میلیون سال پیش»)
۵.۱۰ توصیهها و بهترین شیوهها
✅ از چندین کالیبراسیون فسیلی معتبر استفاده کنید
✅ محدودههای حداقل و حداکثر را با دقت تعیین کنید
✅ هر دو نوع ساعت (سختگیرانه و منعطف) را آزمایش کنید
✅ زنجیرههای MCMC طولانی اجرا کرده و بررسی کنید که به همگرایی رسیدهاند
✅ همیشه بازهی عدم قطعیت (credible intervals) را گزارش کنید
نتیجهگیری بخش ۵
ساعت مولکولی، درختهای فیلوژنتیک را از یک ساختار ایستا به یک جدول زمانی پویا تبدیل میکند.
با ترکیب دادههای ژنتیکی، فسیلها و مدلهای آماری، تاریخگذاری مولکولی به ما امکان داده است درک عمیقتری از تاریخ تکامل و تنوع زیستی پیدا کنیم — از تکامل جانوران باستانی گرفته تا منشأ ویروسها.
با انتخاب درست مدل ساعت، کالیبراسیون دقیق گرهها، و تفسیر محتاطانه عدم قطعیتها، میتوان درختهایی ساخت که نهتنها از نظر ریاضی درست باشند، بلکه از نظر زیستشناسی نیز معنادار باشند.
بخش ۶: فایلوژنومیک و دادههای پرحجم
6.1 فایلوژنومیک چیست؟
فایلوژنومیک (Phylogenomics) ترکیبی از علم تبارشناسی (phylogenetics) و ژنومیک (genomics) است — به معنای استفاده از دادههای ژنومی در مقیاس بالا برای استنتاج روابط تکاملی بین موجودات. برخلاف فایلوژنتیک سنتی که تنها بر یک یا چند ژن تکیه دارد، فایلوژنومیک صدها تا هزاران ناحیه ژنی (loci) را بررسی میکند و تصویری دقیقتر و با وضوح بالاتر از تکامل موجودات ارائه میدهد.
فایلوژنومیک به مشکلاتی که در روشهای قدیمی وجود داشت، پاسخ میدهد، مانند:
-
نسبت پایین سیگنال به نویز در درختهای تکژنی
-
ناهمخوانی بین درخت ژنی و درخت گونهای
-
رخدادهای پیچیدهای مانند مرتبسازی ناقص نسب (Incomplete Lineage Sorting)، هیبریداسیون (Hybridization) و انتقال افقی ژن (Horizontal Gene Transfer)
6.2 ظهور توالییابی پرتوان (High-Throughput)
ظهور تکنولوژیهای جدید توالییابی یا NGS (Next-Generation Sequencing) تحول عظیمی در فایلوژنتیک ایجاد کرده است. این روشها به پژوهشگران اجازه میدهد تا به صورت سریع و مقرونبهصرفه موارد زیر را توالییابی کنند:
-
کل ژنومها
-
ترنسکریپتومها (RNA-seq)
-
ناحیههای هدفگذاریشده از طریق روشهای غنیسازی
-
عناصر بسیار محافظتشده (Ultra-Conserved Elements یا UCEs)
-
اکزومها (مناطق کدکننده پروتئین)
این عصر ژنومیک دقت بیسابقهای را در ساخت درخت زندگی ایجاد کرده است.
6.3 انواع رایج دادههای فایلوژنومیک
|
نوع داده |
توضیح |
کاربرد رایج |
|
ژنوم کامل |
توالی کامل هسته یا اندامکها |
بررسی فایلوژنی عمیق، رخدادهای پیچیده مانند هیبریداسیون |
|
ترنسکریپتومها |
توالییابی mRNA، معمولاً از گونههای غیرمدل |
بررسی تکامل مقایسهای، فایلوژنتیک عملکردی |
|
RAD-seq |
DNA مربوط به سایتهای برشی آنزیمها |
بررسی گونههای نزدیک، تکامل در سطح جمعیت |
|
AHE (Anchored Hybrid Enrichment) |
هدفگیری صدها ناحیه محافظتشده با استفاده از پروب |
ایجاد درختهای دقیق برای جداییهای سطحی و عمیق |
|
UCEs |
نواحی DNA با حفاظت بسیار زیاد |
فایلوژنی در سطوح عمیق |
|
اورتولوگهای تکنسخهای |
ژنهایی با یک همتا در هر گونه |
سیگنال واضح برای درخت گونهای |
6.4 درخت ژنی در برابر درخت گونهای
یکی از مهمترین چالشهای فایلوژنومیک، تمایز بین درختهای ژنی و درخت گونهای است.
-
درخت ژنی: تاریخچه تکاملی یک ژن خاص
-
درخت گونهای: تاریخچه واقعی تکامل گونهها
اما به علت فرآیندهایی مثل:
-
مرتبسازی ناقص نسب (ILS)
-
انتقال افقی ژن (HGT)
-
هیبریداسیون
-
دوبرابر شدن یا حذف ژنها
… درختهای ژنی ممکن است با درخت واقعی گونهای هماهنگ نباشند و باید این ناسازگاریها در مدل لحاظ شوند.
6.5 استنتاج درخت گونهای بر پایه مدلهای همتباری (Coalescent)
برای حل مشکل ناسازگاری درخت ژن و گونه، از مدلهای همتباری (Coalescent models) استفاده میشود. این مدلها دنبال میکنند که چگونه نژادهای ژنی به عقب برمیگردند تا به یک نیا برسند.
روشهای رایج:
-
ASTRAL: استخراج درخت گونهای از مجموعهای از درختهای ژنی بدون ریشه
-
MP-EST و STAR: مدلهایی مشابه بر اساس توپولوژی درختها
-
SVDquartets: به جای درخت کامل، از الگوهای سایتها استفاده میکند
این روشها عدم قطعیت آماری را در نظر میگیرند و از اتصال ساده توالیها قویتر هستند.
6.6 رویکردهای اتصال (Concatenation) و همتباری (Coalescent)
دو راهبرد اصلی برای استنتاج فایلوژنومیک:
1. اتصال (Supermatrix)
-
ترکیب چندین ناحیه ژنی در یک ماتریس تراز شده
-
ساخت یک درخت از تمام سایتها
-
سادگی و قدرت زیاد اما با فرض اینکه همه ژنها تاریخچهای یکسان دارند
2. همتباری (Supertree)
-
ساخت درخت برای هر ژن به صورت جداگانه
-
مدلسازی مرتبسازی ناقص نسب
-
دقیقتر در شرایط پیچیده
بهترین شیوه امروزی: اجرای هر دو روش و مقایسه نتایج، مخصوصاً در نواحی دارای اختلاف نظر.
6.7 تشخیص اورتولوگ و پارالوگ
تشخیص دقیق اورتولوگها (Orthologs) در فایلوژنومیک بسیار مهم است.
-
اورتولوگ: ژنهایی که از یک ژن نیایی از طریق گونهزایی مشتق شدهاند
-
پارالوگ: ژنهایی که از تکرار ژن درون یک ژنوم حاصل شدهاند
استفاده اشتباه از پارالوگها به جای اورتولوگها میتواند درخت را گمراه کند.
ابزارهای رایج:
-
OrthoFinder
-
OMA
-
OrthologID
-
BUSCO (برای ارزیابی کیفیت)
6.8 مواجهه با دادههای ناقص
هیچ مجموعه دادهای کامل نیست. مشکلات رایج:
-
ژنومهای ناقص
-
مونتاژهای با پوشش پایین
-
شکاف در ترازها
-
نبود اورتولوگ در برخی گونهها
راهکارها:
-
استفاده از ابزارهای فیلتر و برش سایتها مانند TrimAl، Gblocks
-
اطمینان از تعداد کافی سایتهای اطلاعاتی در هر ناحیه
-
آزمایش آستانههای کامل بودن داده (مثلاً ۷۰٪، ۹۰٪)
-
استفاده از نرمافزارهایی که با داده ناقص کار میکنند
6.9 روند بیوانفورماتیکی در فایلوژنومیک
یک روند معمول شامل مراحل زیر است:
-
توالی خام → کنترل کیفیت (مانند FastQC، Trimmomatic)
-
مونتاژ → ژنوم یا ترنسکریپتوم (مانند SPAdes، Trinity)
-
تشخیص اورتولوگ → OrthoFinder، BUSCO
-
تراز → MAFFT، PRANK، MUSCLE
-
فیلتر → حذف نواحی بدترازشده
-
انتخاب مدل → ModelFinder در IQ-TREE
-
ساخت درخت → RAxML، IQ-TREE، ASTRAL، BEAST
-
تصویربرداری از درخت → FigTree، iTOL، DensiTree
6.10 چالشهای فایلوژنومیک
|
چالش |
توضیح |
|---|---|
|
ناهمگونی دادهها |
ژنها با نرخ و شرایط متفاوتی تکامل مییابند |
|
ILS و هیبریداسیون |
میتوانند مرزهای گونهای را پنهان کنند |
|
انتقال افقی ژن |
بهویژه در میکروبها رایج است |
|
تغییر در ترکیب بازها |
اگر لحاظ نشود، شکل درخت را تغییر میدهد |
|
تکرار و حذف ژن |
تشخیص اورتولوگ را دشوار میسازد |
رفع این چالشها نیازمند طراحی دقیق آزمایش، مدلهای آماری قوی و فیلتر صحیح داده است.
6.11 فایلوژنومیک در موجودات غیرمدل
فایلوژنومیک اکنون امکان مطالعه موجوداتی را فراهم کرده که قبلاً قابل بررسی نبودند، از جمله:
-
گونههای در خطر انقراض با DNA محدود
-
نمونههای قدیمی یا آسیبدیده
-
تنوع زیستی در مناطق کمتر بررسیشده
-
گونههای پنهان (cryptic species)
کاربردها:
-
اکولوژی
-
ژنتیک حفاظتی
-
زیستشناسی تکاملی رشدی (Evo-Devo)
6.12 کاربردهای فایلوژنومیک
-
حل گرههای عمیق در درخت زندگی (مثلاً منشأ حیوانات یا ریشه یوکاریوتها)
-
درک اشعاعهای سریع (مانند ماهیهای سیکلید، فنچهای داروین)
-
بررسی تکامل خانوادههای ژنی
-
شناسایی درونآمیختگی (introgression) و هیبریداسیونهای باستانی
-
مطالعه تکامل میکروبی و انتقال ژن
نتیجهگیری بخش ۶
فایلوژنومیک دقت و دامنه استنتاجهای فایلوژنتیکی را به شکل چشمگیری افزایش داده است. با استفاده از مجموعه دادههای بزرگ، الگوریتمهای پیشرفته و ابزارهای محاسباتی قدرتمند، پژوهشگران میتوانند درختهایی با وضوح بالا بسازند—even در حضور مشکلاتی مثل مرتبسازی ناقص نسب، تکرار ژن یا هیبریداسیون.
اکنون که توالییابی ارزان و در دسترس شده، فایلوژنومیک فقط مختص موجودات مدل نیست — بلکه دارد دیدگاه ما را نسبت به تنوع حیات در تمام شاخههای درخت زندگی متحول میکند.
بخش ۷: مفاهیم گونه (Species) و فیلوژنی (Phylogeny)
۷.۱ مقدمه: چرا تعریف گونه اهمیت دارد؟
در زیستشناسی تکاملی، تعریف گونه همیشه موضوعی مهم و گاهی چالشبرانگیز بوده است. علم فیلوژنی (تبارزایی) چارچوبی فراهم میکند تا روابط تکاملی بین جانداران بررسی شود. اما نحوهای که گونه را تعریف میکنیم، مستقیماً بر چگونگی ساخت، تفسیر و اعتبارسنجی درختهای فیلوژنتیک تأثیر میگذارد.
گونهها واحدهای پایهای تنوع زیستی، ردهبندی (taxonomy) و حفاظت از طبیعت هستند. با این حال، هیچ تعریف واحد و جهانی برای گونه وجود ندارد. بلکه، مفاهیم مختلفی برای گونه وجود دارد که هر کدام از معیارهای متفاوتی برای طبقهبندی استفاده میکنند.
۷.۲ مفاهیم اصلی گونه
۱. مفهوم زیستی گونه (Biological Species Concept - BSC)
-
تعریف: گونه مجموعهای از جمعیتهای طبیعی است که با هم آمیزش میکنند و از دیگر گروهها بهطور تولیدمثلی جدا شدهاند.
-
ارائهدهنده: ارنست مایر (Ernst Mayr) در سال ۱۹۴۲
-
مزایا:
-
بهخوبی با تصور رایج مردم درباره گونهها در جانداران دارای تولیدمثل جنسی سازگار است.
-
-
محدودیتها:
-
برای موجودات غیرجنسی، فسیلها یا گونههایی که با یکدیگر آمیزش دارند، کاربردی ندارد.
-
در عمل، ارزیابی جدایی تولیدمثلی اغلب دشوار است.
-
۲. مفهوم فیلوژنتیکی گونه (Phylogenetic Species Concept - PSC)
-
تعریف: گونه کوچکترین گروه تکنیا (monophyletic) است که با ویژگیهای اشتراکی و مشتقشده (synapomorphies) قابل شناسایی است.
-
مزایا:
-
برای موجودات جنسی و غیرجنسی قابل استفاده است.
-
بهخوبی با آنالیزهای مولکولی فیلوژنتیک کار میکند.
-
-
محدودیتها:
-
ممکن است منجر به تفکیک بیشازحد گونهها شود (بیشتر از BSC).
-
به کیفیت دادهها و وضوح درخت فیلوژنتیک وابسته است.
-
۳. مفهوم ریختشناسی گونه (Morphological Species Concept)
-
تعریف: گونهها بر اساس ویژگیهای ظاهری قابل مشاهده از هم متمایز میشوند.
-
مزایا:
-
ساده و گسترده قابل استفاده، بهویژه برای فسیلها.
-
-
محدودیتها:
-
ریختشناسی میتواند متغیر و وابسته به محیط باشد.
-
گونههای همریخت (cryptic species) ممکن است نادیده گرفته شوند.
-
۴. مفهوم بومشناختی گونه (Ecological Species Concept)
-
تعریف: گونه مجموعهای از جاندارانی است که یک جایگاه بومشناختی (ecological niche) مشابه را اشغال میکنند.
-
مزایا:
-
بر نقش تطبیقی گونهها و محیط زیست در فرایند گونهزایی تأکید دارد.
-
-
محدودیتها:
-
تعریف جایگاه بومشناختی دقیق دشوار است.
-
ممکن است همپوشانی بین گونههای نزدیک رخ دهد.
-
۵. مفهوم خوشه ژنتیکی/ژنوتیپی گونه (Genetic Cluster Concept)
-
تعریف: گونه بهعنوان خوشههای ژنتیکی متمایز از افراد درون جمعیتها در نظر گرفته میشود.
-
کاربرد: در بومشناسی مولکولی و ژنومیک جمعیتها
-
محدودیتها:
-
نیاز به دادههای گسترده ژنومی دارد.
-
مرزگذاری بین خوشهها ممکن است دلبخواهی باشد.
-
۷.۳ نقش فیلوژنی در تعریف گونهها
آنالیزهای فیلوژنتیک ابزارهای عینی برای تعیین گونهها ارائه میدهند:
-
شناسایی شاخههای تکنیا (monophyletic clades)
-
محاسبه میزان واگرایی ژنتیکی
-
آزمودن مدلهای همتبار شدن (coalescence) برای بررسی خاستگاه مشترک اخیر
ابزارهای نوین فیلوژنتیک میتوانند موارد زیر را شناسایی کنند:
-
گونهزایی پنهان (cryptic speciation)
-
جریان ژنی بین گونهها (introgression)
-
تکامل شبکهای (reticulate evolution) در گروههای پیچیده
برای مثال:
-
مدلهای چندگونهای همتبار شدن (مانند BPP، STACEY) امکان تفکیک گونهها با احتمالسنجی را فراهم میکنند.
-
نرمافزارهای تعیین گونه مانند bPTP، GMYC، Bayes Factor Delimitation درختهای فیلوژنتیک را برای آزمودن فرضیهها مقایسه میکنند.
۷.۴ گونهزایی و درخت زندگی
گونهها ثابت نیستند—آنها با گذشت زمان تکامل یافته و از هم جدا میشوند. فیلوژنی این فرآیند شاخهشدن را به تصویر میکشد، و مفاهیم گونه باید بازتابی از پویایی این فرآیند باشند.
انواع گونهزایی (Modes of Speciation):
|
نوع |
توضیح |
امضای فیلوژنتیک |
|---|---|---|
|
Allopatric |
جدایی جغرافیایی |
گونههای خواهر با همبستگی جغرافیایی |
|
Sympatric |
بدون جدایی جغرافیایی |
معمولاً شناسایی آن دشوار است |
|
Parapatric |
همپوشانی جزیی جغرافیایی |
واگرایی تدریجی در طول یک گرادیان |
|
Peripatric |
جدایی جمعیتی کوچک |
شاخههای طولانی، نشانههای اثر بنیانگذار |
دادههای ژنومی مدرن امکان شناسایی موارد زیر را فراهم کردهاند:
-
واگرایی با جریان ژنی
-
مناطق دورگهزایی (hybrid zones)
-
گونهزایی در مراحل اولیه (incipient species)
۷.۵ چالشهای تعیین گونه بر پایه فیلوژنی
تعیین گونه بر پایه روابط تبارزایی با چالشهایی همراه است:
-
مرتبنشدن کامل تبارها (Incomplete Lineage Sorting - ILS) میتواند جداییهای اخیر را پنهان کند.
-
جریان ژنی بین گونهها، مرزهای گونهای را محو میکند.
-
گونهزایی دورگهای (hybrid speciation) منجر به درختهایی با ساختار شبکهای میشود.
-
سوگیری در نمونهبرداری یا محدودیت در تعداد ژنها ممکن است منجر به شاخههای جعلی شود.
-
پلاستیسیته فنوتیپی (تغییرات ظاهری) و تکامل همگرا میتوانند تفسیرهای ظاهری را گمراه کنند.
بنابراین، امروزه ردهبندی تلفیقی (Integrative Taxonomy) که از فیلوژنی، ریختشناسی، بومشناسی، رفتارشناسی و ژنومیک استفاده میکند، بسیار توصیه میشود.
۷.۶ مطالعات موردی در تعیین گونه با استفاده از فیلوژنی
مطالعه ۱: گونههای پنهان در قورباغهها
قورباغههایی با ظاهر مشابه در آمریکای جنوبی، پیشتر بهعنوان یک گونه طبقهبندی شده بودند. اما آنالیزهای مولکولی (میتوکندریایی و هستهای) تبارهای عمیقاً متفاوتی را نشان داد—هر کدام گونهای جداگانه مطابق PSC و مدلهای همتبار شدن بودند.
مطالعه ۲: فیلهای آفریقایی
در گذشته همه فیلهای آفریقایی بهعنوان یک گونه (Loxodonta africana) در نظر گرفته میشدند. اما آنالیزهای ژنومی نشان داد که در واقع دو شاخه مجزا وجود دارد:
-
فیل دشتی (L. africana)
-
فیل جنگلی (L. cyclotis)
این تفکیک با دادههای ریختشناسی، بومشناسی و تولیدمثلی نیز تأیید شد.
مطالعه ۳: سهرههای داروین (Darwin’s Finches)
در ابتدا بر اساس ریختشناسی بهحدود ۱۴ گونه تقسیم شده بودند. اما مطالعات ژنومی الگوهای پیچیدهای از:
-
مرتبنشدن کامل تبارها
-
جریان ژنی بین گونهها (introgression)
-
تکامل شبکهای
را نشان داد که مرزهای سنتی گونهها را به چالش کشید.
۷.۷ کاربرد مفاهیم فیلوژنتیکی گونه در عمل
در زیستشناسی حفاظت و ارزیابی تنوع زیستی، مفاهیم گونهای مبتنی بر فیلوژنی بیشازپیش مورد استفاده قرار میگیرند:
-
شناسایی واحدهای مهم تکاملی (ESUs)
-
روشن ساختن تمایز تبارها
-
ارائه شواهد برای لیست قرمز IUCN و سیاستگذاریهای حفاظتی
در ردهبندی (taxonomy) نیز کمک میکند به:
-
شناسایی سریعتر مرزهای گونهای
-
ردهبندی بهتر موجودات غیرمدل
-
اولویتبندی برای حفاظت از تبارهای بومی یا نادر
۷.۸ جمعبندی: مفاهیم گونه در دوران فیلوژنومیک
ابزارهای فیلوژنتیک مدرن امکان موارد زیر را فراهم میسازند:
-
تفکیک گونه بر اساس دادههای تجربی و دقیق
-
ترکیب دادههای ژنومی، ریختشناسی و بومشناسی
-
تجزیه و تحلیل تاریخچههای تکاملی پیچیده
هیچ مفهوم واحدی از گونه برای تمام موقعیتها مناسب نیست. بنابراین رویکردی چندمفهومی (pluralistic)—که بر اساس زیستشناسی جاندار و دادههای در دسترس انتخاب میشود—بیشترین کارایی را دارد.
با گسترش روزافزون فیلوژنومیکس (phylogenomics)، درک ما از مفهوم گونه و چگونگی شکلگیری آنها دگرگون شده است. امروزه گونهها نه بهعنوان طبقههایی ثابت، بلکه بهعنوان تبارهایی پویا و در حال تحول در نظر گرفته میشوند.
بخش ۸: کاربردهای فیلوژنی
تحلیل فیلوژنتیک که زمانی تنها بهعنوان ابزاری نظری در زیستشناسی تکاملی شناخته میشد، امروزه به ابزاری حیاتی در بسیاری از شاخههای علمی تبدیل شده است. امروزه در ردهبندی زیستی، زیستشناسی حفاظت، پزشکی، کشاورزی، بومشناسی و بهویژه در اپیدمیولوژی از درختهای فیلوژنتیک استفادههای گستردهای میشود.
در این بخش بررسی میکنیم که چگونه اصول و ساختار درختهای فیلوژنتیک در حل مسائل واقعی و پاسخ به سوالات علمی به کار میروند.
۸.۱ ردهبندی و سیستماتیک (Taxonomy & Systematics)
یکی از ابتداییترین و بنیادیترین کاربردهای فیلوژنی، طبقهبندی و نامگذاری علمی موجودات به شیوهای است که بازتابی از تاریخ تکاملی آنها باشد.
ردهبندی فیلوژنتیک
در سیستماتیک مدرن، روش کلادیستیک (Cladistics) ترجیح داده میشود، که در آن موجودات براساس ویژگیهای اشتراکی مشتقشده (Synapomorphies) گروهبندی میشوند.
در این سیستم، یک تاکسون معتبر باید تکتبار (Monophyletic) باشد؛ یعنی شامل یک نیای مشترک و همهی نوادگان آن باشد.
حل تعارضهای ردهبندی
در مواردی که شکل ظاهری مبهم یا دچار همگرایی تکاملی است، درختهای فیلوژنتیک میتوانند به روشن شدن روابط واقعی کمک کنند.
مثال: مطالعات مولکولی نشان دادند که نهنگها در درون گروه Artiodactyla (پستانداران سمدار زوجسم) قرار دارند و نزدیکترین خویشاوند آنها اسبهای آبی (هیپاپوتاموس) هستند.
بارکد حیات (Barcode of Life)
روش DNA بارکدینگ (مثلاً استفاده از ژن COI در حیوانات) به کمک درختهای فیلوژنتیک میتواند گونهها را حتی از قطعات ناقص یا مراحل نوزادی شناسایی کند.
۸.۲ زیستشناسی حفاظت (Conservation Biology)
فیلوژنی در حفاظت از گونهها کمک میکند تا تبارهای تکاملی متمایز و آسیبپذیر را شناسایی و محافظت کنیم.
مفاهیم کلیدی
-
تمایز تکاملی (Evolutionary Distinctiveness – ED): نشان میدهد یک گونه چقدر در درخت حیات ایزوله است.
-
شاخص EDGE: ترکیبی از تمایز تکاملی و خطر انقراض جهانی برای اولویتبندی در حفاظت.
کاربردها
-
تمرکز بر حفظ گونههایی مانند ماهیهای زندهفسیلی (مثل سیلکانت) یا کلادهای منحصربهفرد با خویشاوندان کم.
-
راهنمایی در برنامههای بازمعرفی (Reintroduction) برای جلوگیری از تداخل ژنتیکی با تبارهای خاص.
-
شناسایی واحدهای تکاملی مهم (ESU) در درون یک گونه با روشهای فیلوژنتیک.
مثال: طوطی پرندهناتوان نیوزیلندی، کاکاپو (Strigops habroptilus)، هم از نظر انقراض در خطر است و هم از نظر تکاملی بسیار منحصربهفرد است.
۸.۳ پزشکی و سلامت عمومی
روشهای فیلوژنتیک در حال تحول پژوهش در بیماریهای عفونی و تصمیمگیری بالینی هستند.
شناسایی و ردیابی پاتوژنها
درختهای فیلوژنتیک میتوانند منشاء، سیر تکامل و انتشار بیماریهایی مانند HIV، آنفلوانزا، ابولا، سارس و کووید-۱۹ را مشخص کنند.
اپیدمیولوژی مولکولی از این درختها برای شناسایی زنجیره انتقال و پرش بینگونهای استفاده میکند.
مقاومت آنتیبیوتیکی (AMR)
فیلوژنومیکس نشان میدهد چگونه ژنهای مقاومت دارویی از طریق انتقال افقی بین باکتریها پخش میشوند.
همچنین میتوان با آن تکامل مقاومت دارویی را پیشبینی کرد.
فیلوژنی سرطان
تومورها ساختار کلونی دارند و در طول زمان تکامل مییابند. محققان با استفاده از فیلوژنی درونتوموری میتوانند:
-
ترتیب جهشها را بازسازی کنند
-
مسیرهای متاستاز را پیشبینی نمایند
-
درمانهای هدفمند را بهتر طراحی کنند
توسعه واکسن
درختهای فیلوژنتیک در پیشبینی تغییرات آنتیژنی آنفلوانزا مؤثرند و در انتخاب سویه واکسن سالانه کمک میکنند.
در مورد SARS-CoV-2، درختهای جهانی کمک کردند تا گونههایی مانند آلفا، دلتا و اُمیکرون پیگیری شوند.
۸.۴ کشاورزی و علوم گیاهی
درختهای فیلوژنتیک ابزار مهمی در زمینههای زیر هستند:
-
اصلاح نژاد محصولات کشاورزی
-
شناسایی عوامل بیماریزا
-
مطالعه فرآیند اهلیسازی گیاهان
اهلیسازی گیاهان
درختها میتوانند نیاکان وحشی گیاهان اهلیشده مانند گندم، ذرت و برنج را شناسایی کنند.
کمک به حفظ منابع ژنتیکی (Germplasm) و نقشهبرداری صفات زراعی.
مدیریت آفات و بیماریها
با استفاده از درختها میتوان گسترش آفات و مقاومت آنها را دنبال کرد.
مثال: درخت فیلوژنتیک قارچ Phytophthora infestans، عامل بلایت سیبزمینی، در پیشبینی شیوعهای آینده نقش دارد.
تراریختهها و فیلوژنی
درختهای مبتنی بر DNA میتوانند خلوص ژنتیکی را تأیید کنند، تلاقیهای ناخواسته را شناسایی کنند، و جریان ژنی بین محصولات تراریخته و گونههای وحشی را بررسی کنند.
۸.۵ بومشناسی و ساختار جوامع زیستی
درختهای فیلوژنتیک باعث درک بهتر همزیستی و تعامل بین گونهها در اکوسیستمها میشوند.
تنوع فیلوژنتیک (PD)
معیاری برای اندازهگیری تنوع زیستی بر اساس طول شاخهها و روابط تکاملی بهجای فقط تعداد گونهها.
فیلوژنتیک جوامع (Community Phylogenetics)
بررسی میکند آیا یک جامعه زیستی از:
-
گونههای نزدیک به هم (Clustering): نشاندهنده فیلتر شدن محیطی
-
گونههای دور از هم (Overdispersion): نشاندهنده رقابت و حذف گونههای مشابه
ثبات نیچ (Niche Conservatism)
بررسی این که آیا گونههای خویشاوند نیچهای مشابهی را در طول زمان حفظ میکنند یا نه.
۸.۶ پزشکی قانونی و حقوقی
درختهای فیلوژنتیک اکنون در دادگاهها و تحقیقات جنایی نیز کاربرد دارند:
-
ردیابی انتقال بیماری: مثلاً بررسی اینکه آیا HIV عمداً از فردی به فرد دیگر منتقل شده یا نه.
-
ردیابی قاچاق حیاتوحش: با استفاده از دادههای ژنتیکی میتوان منشاء گونههای قاچاقشده را مشخص کرد.
-
بررسی حملات بیوتروریستی: شناسایی سویههای منشاء سلاحهای زیستی بالقوه مانند باسیلوس آنتراسیس (عامل سیاهزخم).
۸.۷ تکامل زبان و فیلوژنی فرهنگی
شگفتانگیز است که روشهای فیلوژنتیک حتی در سامانههای غیر زیستی نیز کاربرد یافتهاند:
-
زبانشناسی تاریخی: بازسازی درخت زبانها مانند زبانهای هندواروپایی
-
تکامل فرهنگی: مدلسازی تکامل ابزار، مراسم، و سنتها
-
فیلوژنی نسخههای خطی: بازسازی تغییرات و انتقال متون باستانی
این کاربردها بر پایه همان اصول کلادیستیک و استنباط بیزی هستند که در فیلوژنی زیستی استفاده میشود.
۸.۸ محیطزیست و مطالعات متاژنومیک
در بررسی تنوع میکروبی در محیطهای مختلف مانند خاک، اقیانوس یا روده انسان، ابزارهای فیلوژنتیک ضروری هستند.
متابارکدینگ و eDNA
با قرار دادن توالیها در درختهای فیلوژنتیک میتوان:
-
گونههای ناشناس را شناسایی کرد
-
تغییرات اکوسیستم را بررسی کرد
-
کلادهای جدید را کشف کرد
تحقیقات میکروبیوم
درختها به ما کمک میکنند تا:
-
همتکامل میزبان و میکروبهایش را ردیابی کنیم
-
پاسخ جوامع میکروبی به رژیم غذایی، بیماری و آنتیبیوتیکها را بفهمیم
۸.۹ زیستشناسی سنتتیک و زیستفناوری
مهندسان زیستی از فیلوژنی برای اهداف زیر استفاده میکنند:
-
ردیابی منشاء ژنهای عملکردی
-
طراحی آنزیمهای کایمریک یا مسیرهای زیستی مصنوعی
-
پیشبینی عملکرد پروتئینها بر اساس حفاظت تکاملی
مثال: طراحی انواع جدید سیستمهای CRISPR و آنزیمهای Cas، از طریق بررسی فیلوژنتیک سیستمهای ایمنی باکتریها انجام شده است.
۸.۱۰ خلاصه
فیلوژنی دیگر محدود به سیستماتیک زیستی نیست. امروزه این علم در حوزههایی متنوع مانند:
-
پزشکی انسانی و اپیدمیولوژی
-
حفاظت از محیط زیست و تنوع زیستی
-
کشاورزی و امنیت غذایی
-
پزشکی قانونی و مطالعات فرهنگی
نقشی اساسی دارد. با افزایش تولید دادهها، قدرت تحلیل فیلوژنتیک نیز رشد میکند و در کشف الگوها، آزمون فرضیهها و هدایت اقدامات کاربردی اهمیت بالایی یافته است.